|
|
The Brain Simulation Platform "Live Papers" |
|
|
The Brain Simulation Platform "Live Papers" |
Authors: Kokh DB1, Kaufmann T1,2, Kister B1,3, Wade RC1,3,4,5.
Author information: 1 Molecular and Cellular Modeling Group, Heidelberg Institute for Theoretical Studies (HITS), Heidelberg, Germany. 2 Department of Physics, Heidelberg University, Heidelberg, Germany. 3 Department of Biosciences, Heidelberg University, Heidelberg, Germany. 4 Zentrum für Molekulare Biologie der Universität Heidelberg, DKFZ-ZMBH Alliance, Heidelberg University, Heidelberg, Germany. 5 Interdisciplinary Center for Scientific Computing (IWR), Heidelberg University, Heidelberg, Germany.
Corresponding author: Rebecca C. Wade(Rebecca.Wade@h-its.org) Daria B. Kokh(Daria.Kokh@h-its.org)
Journal: Frontiers in Molecular Bioscience
Download Url: https://doi.org/10.3389/fmolb.2019.00036
Citation: Kokh DB, Kaufmann T, Kister B, Wade RC(2019) Machine Learning Analysis of τRAMD Trajectories to Decipher Molecular Determinants of Drug-Target Residence Times Front. Mol. Biosci. (2019) .
DOI: https://doi.org/10.3389/fmolb.2019.00036
Licence: the Creative Commons Attribution (CC BY) license applies for all files. Under this Open Access license anyone may copy, distribute, or reuse the files as long as the authors and the original source are properly cited.
Here you can find the data used and generated in this work, along with the visualization of the main results produced.
Representative 3D experimental structural data:
| Complex of HSP90 with a resorcinol-type compound bound to loop-type conformation (shown in Fig.1A) |
PDB 5J2X |
archive Download |
3d_rotation View |
| Complex of HSP90 with a recorcinol-type compound bound to the helix-type conformation of the binding site (shown in Fig.1B) |
PDB 5J9X |
archive Download |
3d_rotation View |
Complex of HSP90 with a tricyclic compound bound to helix-type conformation of the binding site (shown in Fig.1D) |
PDB 2YKI |
archive Download |
3d_rotation View |
1D/2D structure data of molecular compound used in the study:
| 1D structures (SMILES format): |
archive Download |
2D structures (sdf format): |
archive Download |
Kinetic rates of protein-ligand binding:
| Experimental Kinetics data |
archive Download |
References:
[1] Amaral, M., Kokh, D. B., Bomke, J., Wegener, A., Buchstaller, H. P., Eggenweiler, H. M., et al. (2017). Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat. Commun. 8, 2276.
[2] Kokh, D., Amaral, M., Bomke, J., Graedler, U., Musil, D., Buchstaller, H., et al. (2018). Estimation of Drug-Target Residence Times by τ- Random Acceleration Molecular Dynamics Simulations. J. Chem. Theory Comput 14, 3859–3869.
[3] Schuetz, D. A., Richter, L., Grandits, M., Graedler, U., Buchstaller, H., Eggenweiler, H., et al. (2018b). Ligand Desolvation steers on-rate and impacts Drug Residence Time of Heat shock protein 90 ( Hsp90 ) Inhibitors. J. Med. Chem. 90, 4397–4411.
Exploring clustering of dissociation trajectories by the overall protein-ligand contact similarity
|
All compounds (Fig. 5) |

Claster composition |
Helix-binding compounds (PDB 5J9X) |
3d_rotation View |
Loop-binding compounds (PDB 5J2X) |
3d_rotation View |
|
Indazole-type compounds (Fig. 6) |
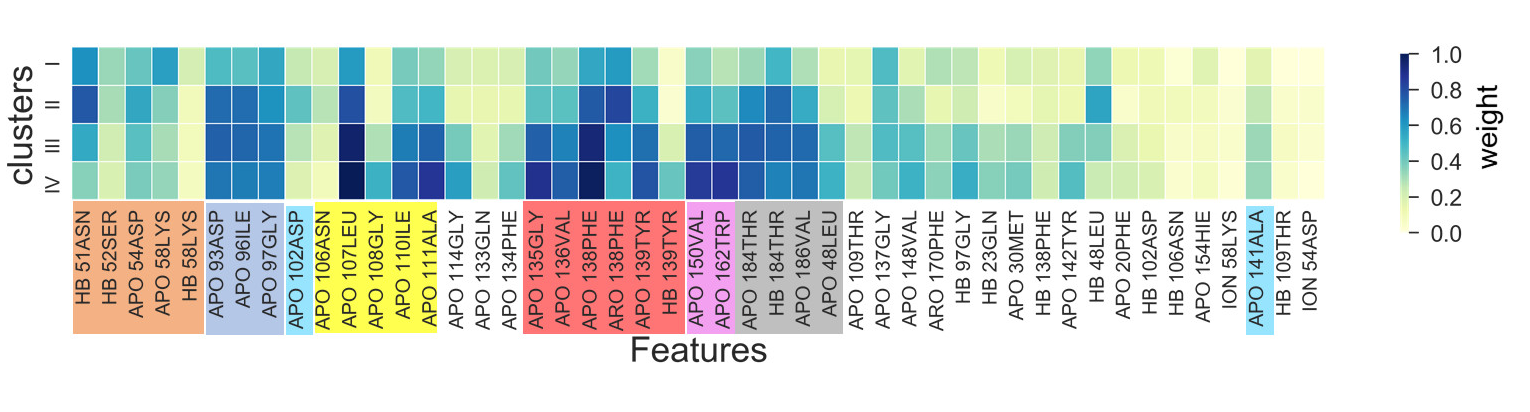
Claster composition |
Indazole-type compounds (PDB 5LNZ) |
3d_rotation View |
Exploring Regression Models for prediction of unbinding rates
| All compounds | Model evaluation (Fig.8) |
timeline View |
Residue contribution to the Linear Regression model |
timeline View |
Residues with a large contribution to the LR model |
3d_rotation View |
| Outliers excluded | Model evaluation (Fig.8) |
timeline View |
Residue contribution to the Linear Regression model |
timeline View |
Residues with a large contribution to the LR model |
3d_rotation View |
| All compounds | Model evaluation |
timeline View |
Residue contribution to the Linear Regression model |
timeline View |
Position of residues with a large contribution to Linear Regression model |
3d_rotation View |
| All compounds | Model evaluation (Fig.8) |
timeline View |
Residue contribution to the Linear Regression model |
timeline View |
Position of residues with a large contribution to Linear Regression model |
3d_rotation View |
Complete archive incuding source codes and data:
| Zenodo archive |
archive Open |